Columbia University Irving Medical Center
Transforming human health by driving discovery, advancing care and educating leaders

Our Schools
Advancing Care
News
- April 25, 2024
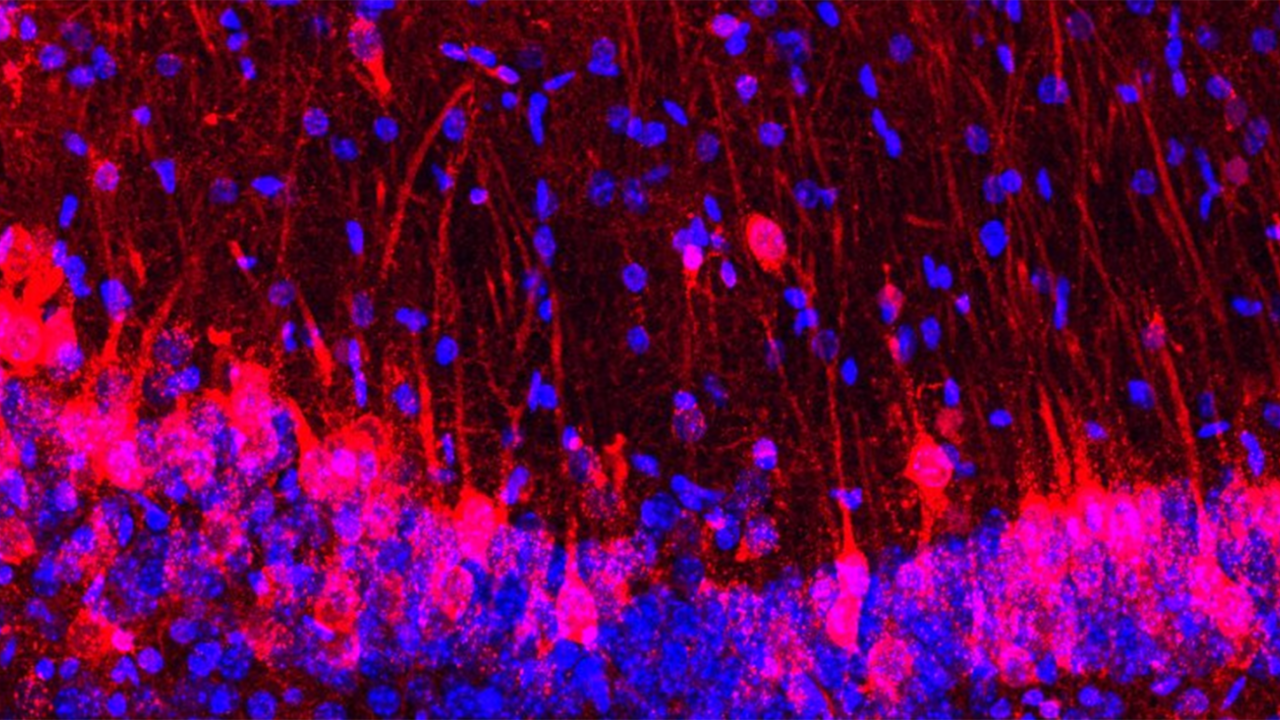
Mice lacking an olfactory system have had their sense of smell restored with rat neurons, the first time scientists have successfully integrated the sensory apparatus of one species into another.
Topic
- April 30, 2024
Adana Llanos, PhD, MPH, confronts racial inequities in breast cancer and unveils the hidden impacts of chemical exposures from personal care products.
Topic
- April 25, 2024
Many surgeons remove the bursa when repairing rotator cuff injuries, but a new animal study suggests that the small tissue helps with healing.
Topic
- May 1, 2024
Arthur G. Palmer and Oliver Hobert of the Department of Biochemistry & Molecular Biophysics were selected in recognition of their distinguished and continuing achievements in research.
Topic
- April 30, 2024
Columbia researchers have found that cells inside clogged arteries have cancer-like properties that aggravate atherosclerosis, and anticancer drugs could be a new treatment.
Topic




Our Community

Learn more about our role in serving the Northern Manhattan community of Washington Heights, Inwood, and Harlem.
Diversity, Equity, and Inclusion

At CUIMC, we are committed to providing culturally inclusive medical education, research, and clinical care.
Events
- Wednesday, May 1, 2024 to Saturday, May 4, 2024All day
- Wednesday, May 1, 2024 to Saturday, May 4, 20248:00 AM to 5:30 PM
- Thursday, May 2, 20249:30 AM to 12:00 PM
- Thursday, May 2, 202411:30 AM to 12:30 PM